
Charge Transfer in Peptide Nucleic Acid (PNA)

Collaborators: Prof. Catalina Achim (Carnegie Mellon University), Dr. Marcela Madrid (Pittsburgh Supercomputing Center) Prof. Eric Borguet (Temple University), Prof. David Beratan (Duke University)
This project is made possible by a grant from the National Science Foundation Collaborative Research in Chemistry (NSF-CRC) initiative.

Group Members: Kathy Davis, Xing Yin , Emil Wierzbinski
Peptide nucleic acid (PNA) is a non-natural DNA analogue that forms helical duplexes and obeys Watson-Crick base-pairing rules. PNA’s neutral, pseudo-peptide amide backbone, resistance to hydrolytic enzymes, and higher affinity for complementary nucleic acids makes it a desirable scaffold for biomimetic devices and bioelectronics. Charge transfer through these oligomers is being studied using cyclic voltammetry (Pitt), scanning probe microscopy (Pitt/Temple), time-correlated single-photon counting (Pitt), and is also modeled theoretically (Duke/PSC).
We have recently examined self-assembled monolayers (SAMs) of PNA oligomers (see figure below) to study the effect of nucleobase identity on the standard heterogeneous rate constant, k0. For both single-stranded and double-stranded PNA, the presence of guanine, which is the most easily oxidized nucleobase, accelerates charge transfer. While the charge transport mechanism in single-stranded PNA was shown to be hole-mediated superexchange (J. Am. Chem. Soc. 2009, 131, 6498-6507), the charge transport for double-stranded PNA likely occurs by a mixture of coherent superexchange and incoherent hopping.
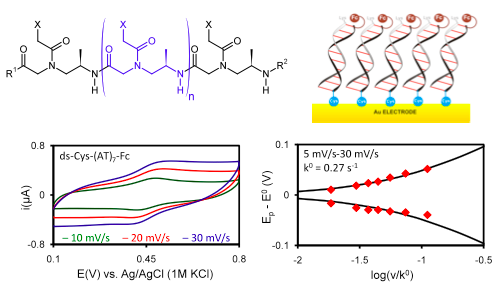
Upper left: Structure of a single-stranded PNA oligomer. X represents the nucleobases G, C, A, or T; R1 and R2 denote functional groups placed at the C- and N-terminus, respectively, of the PNA backbone. Upper right: Cartoon schematic of PNA oligomers with C-terminal cysteine and N-terminal ferrocene groups self-assembled on a gold electrode surface. Lower left: cyclic voltammograms of a PNA SAM composed of double-stranded Cys-(AT)7-Fc PNA. Lower right: A plot of the position of the oxidation (Ep – E0 > 0) and reduction (Ep – E0 < 0) peaks with increasing (normalized) scan rate is fitted with a curve derived from Marcus theory to determine the charge transfer rate constant, k0.
Currently, we have begun to investigate the charge transfer properties of PNA oligomers containing non-natural nucleobase analogues. Our collaborators in the Achim group at Carnegie Mellon University are synthesizing single-stranded PNA oligomers in which one or more bases are selectively replaced by ligands for metal ions, and a duplex is formed with a complementary strand of PNA containing ligands in matching positions. These duplexes are capable of binding metal Co2+, Ni2+, Zn2+, or Cu2+ metal ions. We are asking several important questions about charge transfer through PNA with these systems. Does charge transfer occur by hopping or by tunneling? Does the mechanism change with the position of the metal ion? How does the rate and mechanism change with the position of the metal ion relative to the ferrocene? Can the identity and redox potential of the metal ion change the mechanism? Clarifying these questions will aid in the eventual development of efficient devices using metallized nucleic acids.
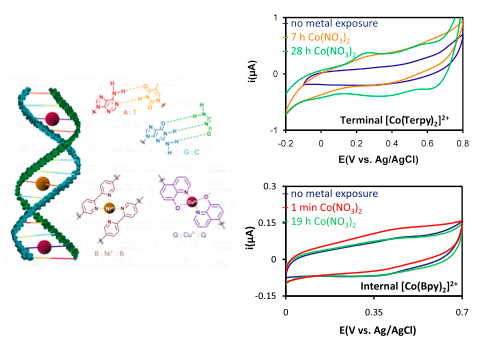
Left: Cartoon schematic of a PNA sequence containing multiple metal ions. Metal ligands, such as bipyridine (B) or 8-hydroxyquinoline, inside the PNA duplex bind metal ions, such as Ni2+ or Cu2+. Upper right: Co2+ binds to a SAM composed of terpyridine (Terpy)-terminated PNA duplexes over a period of 28 hours. Lower right: Co2+ binds to a SAM composed of ferrocene-terminated PNA duplexes having an internal, complementary pair of bipyridine (Bpy) ligands over a period of 19 hours.