|
|
Research Interests
Accomodation of excess charge by water clusters
Excess electrons and protons in water are engaged in a wide range of important chemical,
biological, and geochemical processes. Our group has been especially interested in
understanding how these charged particles are accommodated by the water networks. Much
of our work in this area is in collaboration with the Johnson group at Yale, which uses
vibrational predissociation spectroscopy as a probe of the structure of the clusters.
The resulting spectra tend to be highly anharmonic, providing a significant challenge
to theory. Our group has been engaged in the development of model Hamiltonian approaches
to characterize excess electrons in water and to understand the trends in the OH stretch
spectra of protonated water clusters.
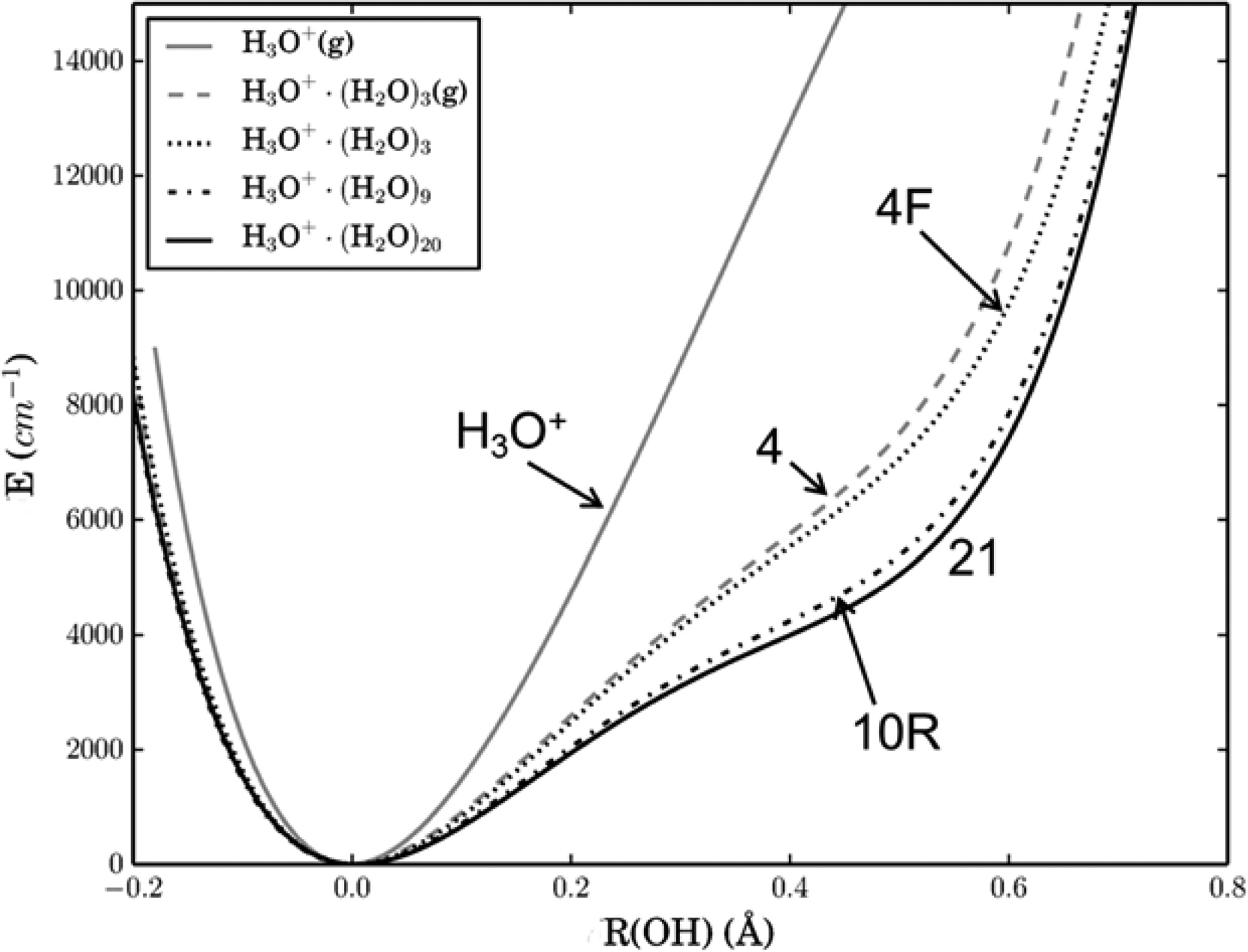
Figure 1: Potential energy of the OH bond of H3O+ as it is successively solvated by hydration shells to form H(H2O)21 +. This plot shows that hydration results in increased anharmonicity in the bond, which was shown to be due to the electric fields of the hydrating water molecules.
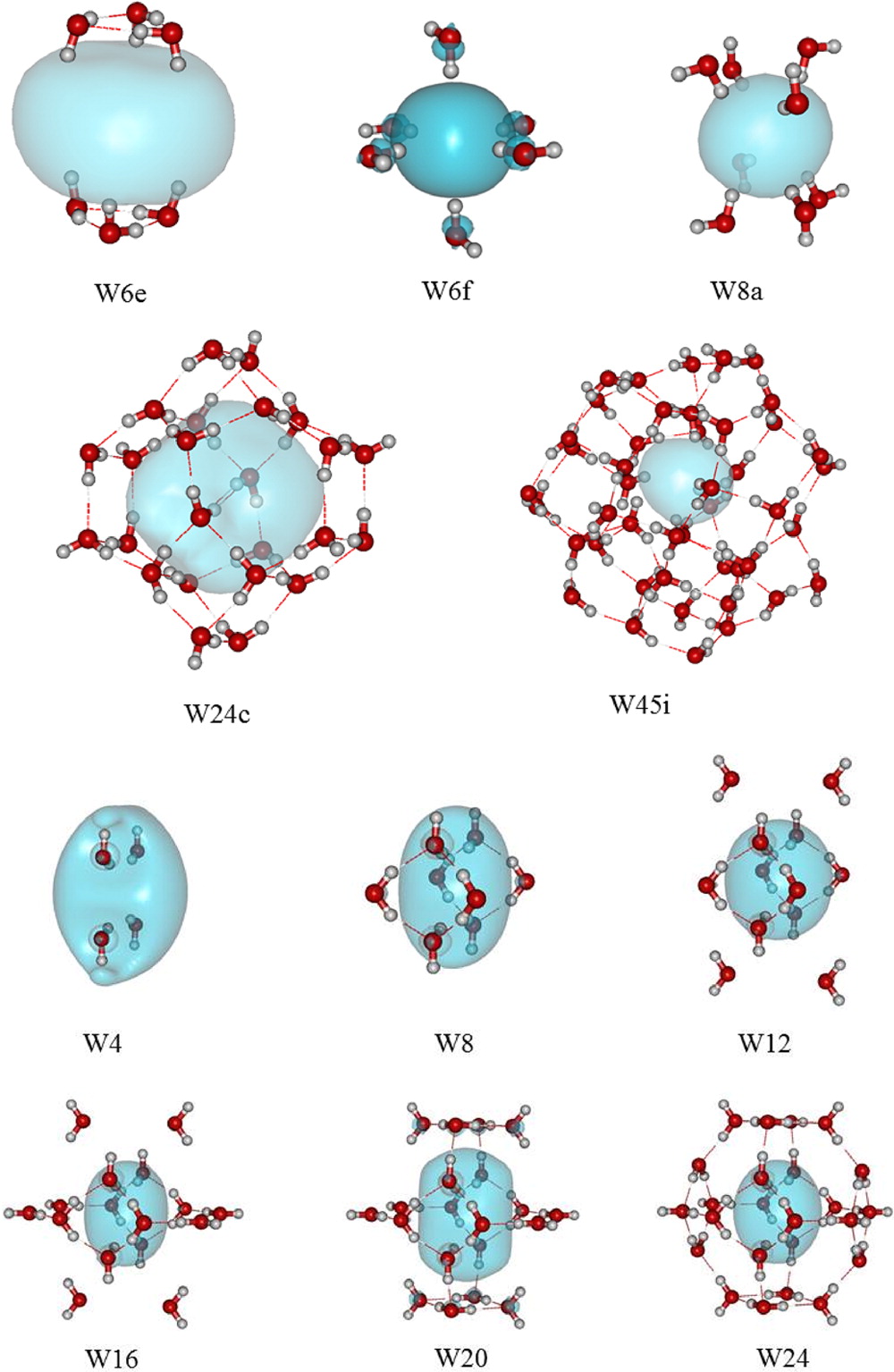
Figure 3: Bound states of various water clusters with an excess electron in the interior cavity. The electron density depicted encloses 90% of the charge density.
Figure 1: Potential energy of the OH bond of H3O+ as it is successively solvated by hydration shells to form H(H2O)21 +. This plot shows that hydration results in increased anharmonicity in the bond, which was shown to be due to the electric fields of the hydrating water molecules.
Figure 3: Bound states of various water clusters with an excess electron in the interior cavity. The electron density depicted encloses 90% of the charge density.
Long-range correlation effects
We are engaged in developing methods to describe long-range correlation effects in molecules,
clusters, and at surfaces. This work includes extensions of the dispersion-correlated
atomic potential (DCACP) procedure of Rothlesberger and co-workers, and the use of quantum
Drude oscillators to describe long-range correlation effects between excess electrons and
molecules and clusters.
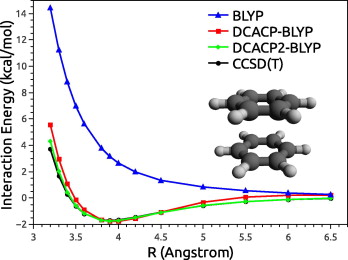
Figure 3: Interaction energy of the sandwich form of the benzene dimer as a function of the
separation between the two monomers. This plot shows that the DCACP2 method is very close
to the CCSD(T) gold standard.
Quantum Monte Carlo methods
Coupled cluster singles plus doubles with perturbative triples [CCSD(T)] is the
"gold standard" of computational chemistry. However, CCSD(T) calculations scale as N7 with
the number of electrons and also require the use of very large basis sets to achieve
conveyance. On the other hand, the diffusion Monte Carlo (DMC) method scales as N3, albeit
with a large prefactor. The DMC method is highly parallel and can be run over tens of
thousands of CPU cores enabling calculation of accurate energies for systems for which
large basis set CCSD(T) calculations are not feasible. The main approximation of DMC
calculations is the fixed-node approximation, which is made to maintain fermionic character
of the wavefunction. Our research is focused on the development of improved nodal
approximations via the use of multiconfigurational trial functions.
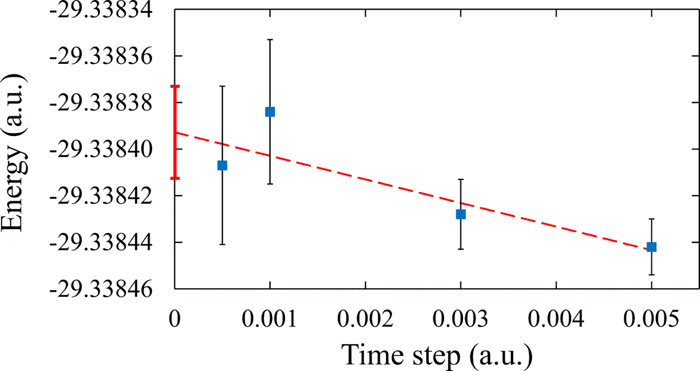
Figure 4: Extrapolation to zero time step of the DMC energy of the Be dimer at the equilibrium bond length of 2.453 603 Å. The calculations were based on the CAS(4,16)/cc-pVQZ-g trial function and used a 0.001 threshold on the CI coefficients.
Sustainability
We are using computational methods to address a range of problems relevant to clean energy
and sustainability. These include modeling heat transport in methane hydrate and other
hydrates and elucidation of the role of water in the uptake of CO2 by clays. In these
studies, we are using classical Monte Carlo and molecular dynamics simulation methods
with classical force fields.
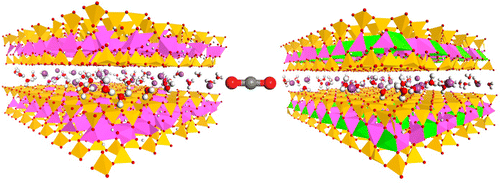
Figure 5: Multiphase Gibbs ensemble Monte Carlo simulations of Na-montmorillonite and Na-beidellite interacting with CO2 and H2O at pressure and temperature conditions relevant for geological storage aquafers.
Other research interests
- Thermodynamics and dynamics of finite systems.
- Potential energy landscapes of biomolecules.
- Dynamics, spectroscopy, and thermal properties of clathrate hydrates.
- Chemistry of adsorbed molecules on metal, semiconductor, and metal oxide surfaces.
- Improved force fields for simulations of complex systems.
- Parallel computing and high performance computing in chemistry and materials science.