
CONFORMATIONAL ANALYSIS OF KETAMINE
Introduction
After PCP (I) was made a Schedule II drug, legitimate
pharmaceutical labs
were trying to modify the structure in order to create
a drug with the useful anesthetic properties of PCP but lacking its peculiar
pyschic effects (1).
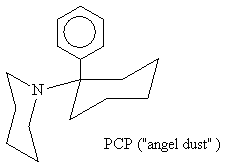
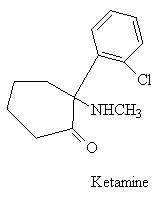
The most successful replacement, to date, is ketamine (II- ketalar, special K, vitamin K) which is a prescription anesthetic . It induces a state of sedation and amnesia in which the patient is still conscious but dissociated from the environment, immobile, and unresponsive to pain. However, psychic disturbances like those from PCP (e.g., hallucinations) can accompany recovery.
Links to ketamine references:
http://area51.upsu.plym.ac.uk/~harl/ketamine.html
http://www.globalideasbank.org/socinv/SIC-150.HTML
Ketamine is known to bind to the NMDA receptor PCP1 site
where PCP
binds:
http: //www.frognet.net/dxm/neuropharm.html
Is there an endogeneous ligand that binds to the PCP1 site? The most promising candidates for the NMDA PCP1 site seems to be a series of peptides (see above reference link).
At this time, it is unknown how ketamine binds to the PCP1 site.
We were interested in carrying out a series of molecular mechanics calculations on both free base and protonated ketamine in an effort to develop a feel for the biologicaly -active conformation of ketamine which binds to the receptor site.
In our calculations we have include possible conformations for PCP (free base and protonated) for comparison.
Calculations were carried out with the Tripos molecular mechanics force-field contained in "Alchemy III". No influence of solvent was icluded, i.e., calculations are done "in vacuo".
Possible conformations for PCP:
Free-base/ phenyl group equatorial/ piperidine ring axial scenario
Calculated Heat of formation is 10.74 Kcal/ Mole. Both piperidine and cyclohexane rings are in the chair conformation.
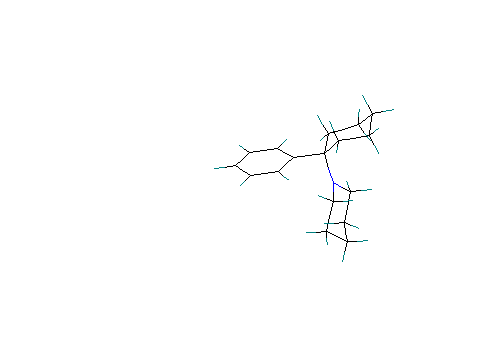
Scenario # 2: phenyl group axial and piperidine ring equatorial
Heat of formation (calculated) at 10.01Kcal/ mole. Both alicyclic rings are in the chair conformation.
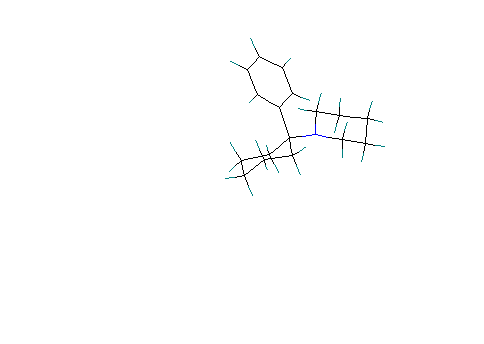
Scenario #3. Phenyl group axial and piperidine ring equatorial.
Nitrogen is protonated.
Notice that protonation of the nitrogen (based on the PKb of piperdine, about 75% of the protonated form should occur in serum) causes the ring to assume the boat conformation. Calculated Heat of formation = 18.75 Kcal/mole :
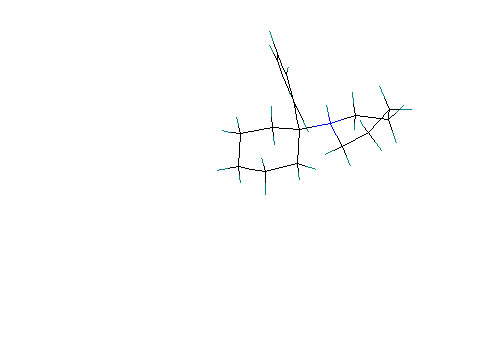
Scenario #4: phenyl group equatorial and protonated piperidine
ring is axial:
The protonated piperidine retains the chair conformation
with the phenyl group equatorial. The calculated Heat of formation = 25.3
Kcal/ mole indicating that this conformation is significantly less stable
(about 7.0 Kcal./mole difference) than the conformation presented in scenario
#3.
If the piperidine ring remains protonated at the PCP receptor site, there is a significant chance that binding is occuring through the conformation shown in scenario #3 with protonated piperidine in the boat conformation.
The same calculations will now be carried out on the ketamine structure .
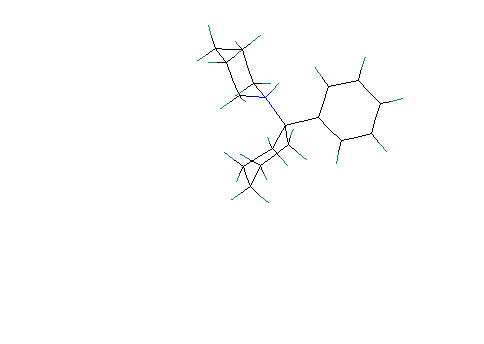
Scenario # 5 Phenyl group axial and amino group equatorial. Free base form:
Calculated Heat of formation = 5.5 Kcal/ mole . Cyclohexanone
is in a chair conformation.
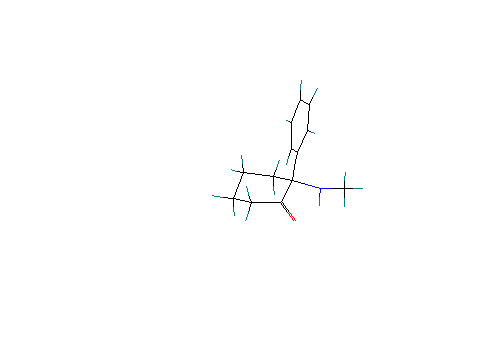
Scenario # 6: Phenyl group equatorial and amino group (free base) axial:
With a calculated Heat of formation = 5.2 Kcal/ mole,
there is no significant
Difference in the energetics of these two free-base conformations.
Will protonation of the amine significantly cause a change in conformational preference? With the presence of a carbonyl group, intramolecular H- bonding is a definite possibility.
Calculations were now carried out on the protonated forms
using the MMX force-field contained in " PC Model" (Serena Software, Bloomington,
Indiana). This force-field, developed by Gajewski and Gilbert, allows intramolecular
H- bonding to be included in the energetics calculations.
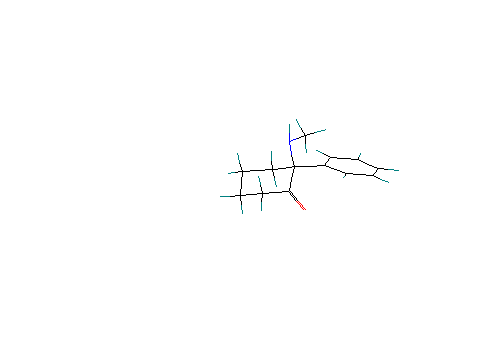
Scenario #7. Protonated ketamine with phenyl group axial. No H bond considered between the amine and the carbonyl group:
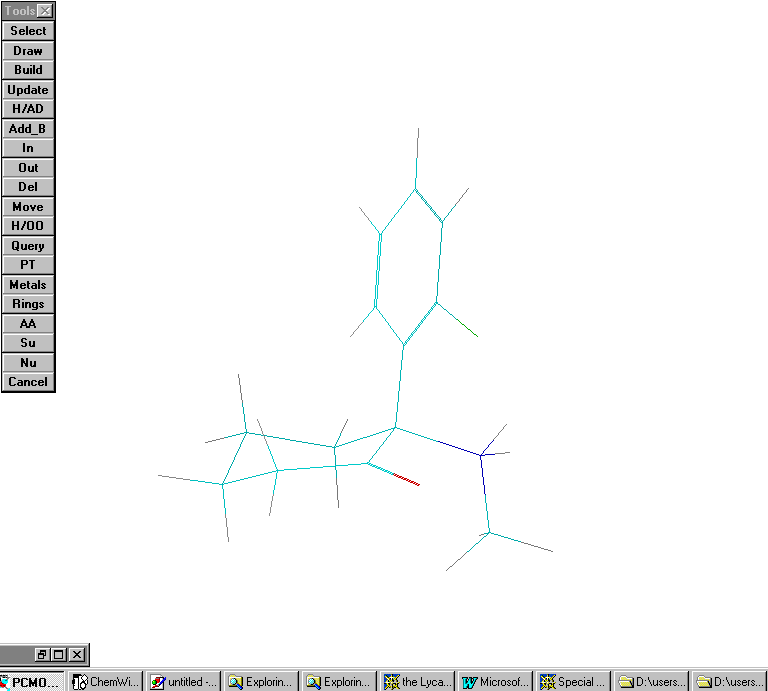
Calculated Heat of formation = 153.40 Kcal/ mole. Cyclohexanone ring is in a chair conformation.
Scenario #8: phenyl group axial and protonated nitrogen forms intramolecular H bond with the carbonyl:
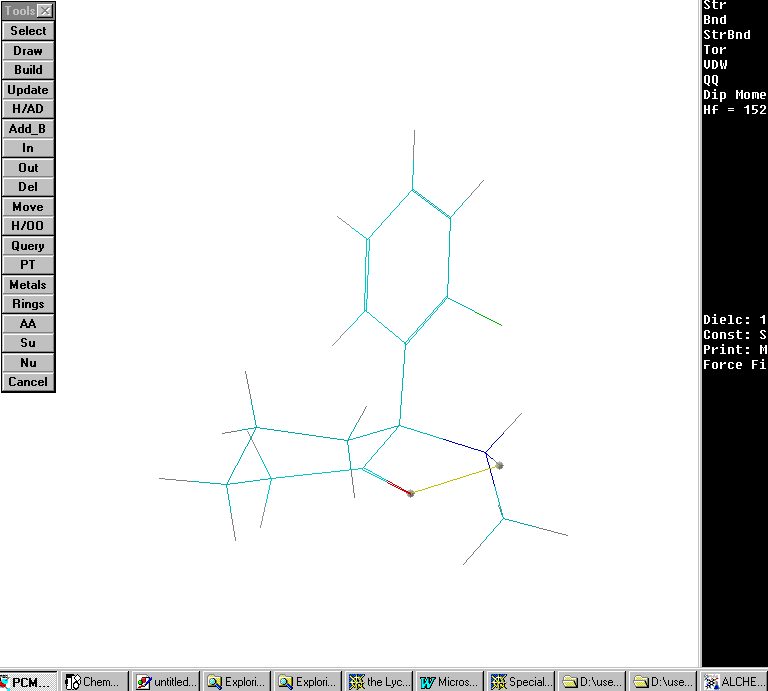
Calculated Hf= 152.8 Kcal/ mole. The interatomic distance between proton and carbonyl held at 1.9 Angstroms for the intramolecular H bond. No significant difference in the energetics results (comparing scenario # 7 and 8).
Scenario #9: phenyl group equatorial. No Intra- H bond considered
Protonated amine:
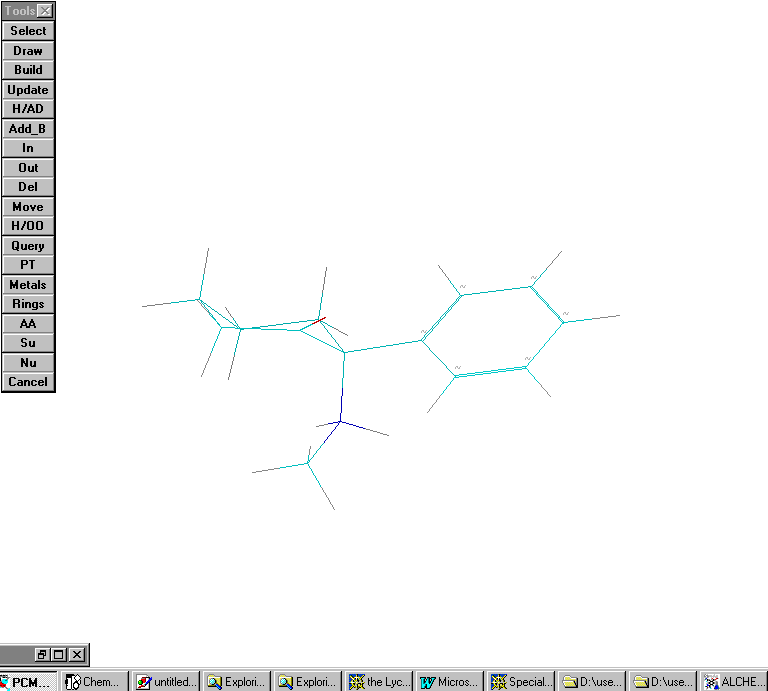
Calculated Hf= 135 Kcal/ mole; significant difference
in energetics results.
Scenario # 10: intramolecular H bonding now involved:
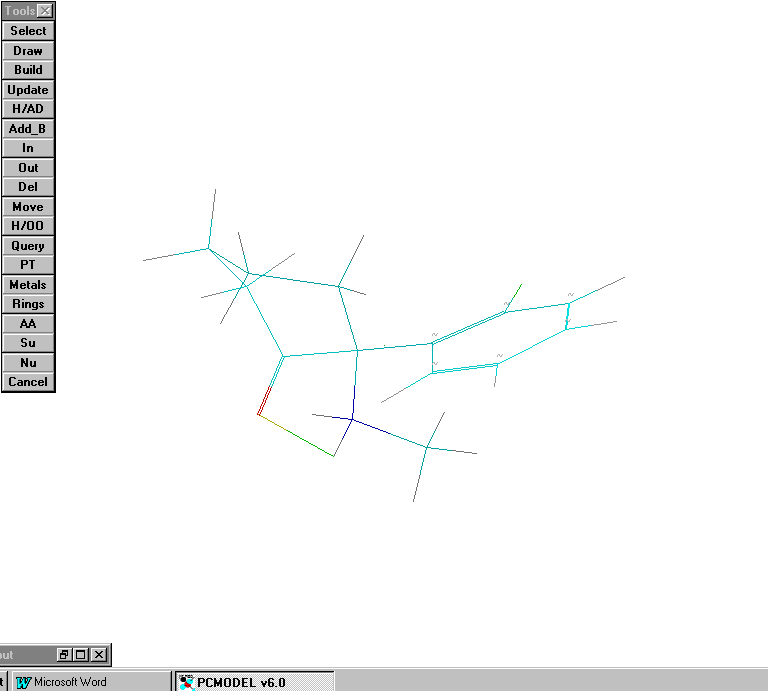
Calculated Heat of formation now = 127 Kcal/ mole. This is a signicant savings of about 4 Kcal vs. the conformation in scenario # 9. It is quite possible that this is the bio-active conformation involved in binding to the PCP1 receptor-site.
How do you go about proving this? You design and synthesize a rigid compound which would superimpose with the shape shown in scenario #
10.
Interested in this type of research? Look into a career in medicinal chemistry and computer- aided drug design/ analysis. Related career choices could also be neurochemistry, molecular pharmacology, and pharmacognosy/ natural products.
We hope you enjoyed this page. Please visit our related
web- site dealing with the conformational analysis of
LSD!
contact us! Soriano+@pitt.edu