| front |1 |2 |3 |4 |5 |6 |7 |8 |9 |10 |11 |12 |13 |14 |15 |16 |17 |18 |19 |20 |21 |22 |23 |24 |25 |26 |27 |28 |29 |30 |31 |32 |33 |34 |35 |36 |37 |38 |39 |40 |41 |42 |43 |44 |45 |46 |47 |48 |49 |50 |51 |52 |53 |54 |55|56 |57 |58 |59 |60 |61 |review |
 |
Finishing
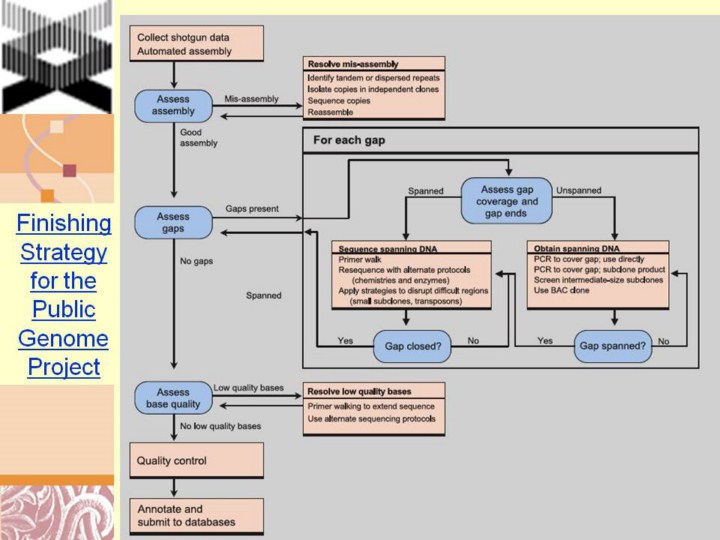
Finishing the sequence of clones Sequencing of large-insert clones began with production of an initial assembly based on shotgun sequence data. For a typical BAC clone, we generated 6–10-fold coverage in paired end-sequences from random, small-insert (2–4 kb) plasmid clones and used computer programs (P. Green, unpublished and see ref. 68) to assemble the data into sequence contigs connected by linking information. Each base was assigned a quality score reflecting its predicted accuracy, based on the underlying shotgun sequence data. The assembly typically had gaps and lowquality regions, with the number varying greatly across clones. These regions are highly enriched in sequences that are difficult to clone or sequence and thus are not represented even after deep (6–10-fold) coverage with random reads. (These regions are also poorly covered by whole-genome shotgun strategies69.) The finishing phase converted this draft assembly into a high-quality continuous sequence by obtaining directed information. It involved iterative cycles of computational analysis and laboratory work. Box 2 Fig. 1 shows a simplified flowchart. The first step was to inspect the draft assembly for evidence of mis-assembly, arising from inappropriate merger of repeated sequences. Such evidence would include inconsistent patterns of linking among contigs, regions with unusually high coverage in sequence reads and bases with ‘high-quality discrepancies’ among the underlying sequence reads. In general, sequence assembly is more straightforward for the clone-based hierarchical shotgun strategy than for the whole-genome shotgun strategy, because the use of clones avoids problems arising from polymorphism and from different copies of repeated regions elsewhere in the genome. Most clones passed assembly inspection, but some failed due to the presence of very similar local dispersed, tandem or inverted repeats. Careful inspection could resolve the problem in some cases, but specific strategies had to be devised in other cases. One approach was to isolate distinct copies of the repeat in subclones of intermediate size (10-kb plasmids or fosmids) and sequence these subclones. Box 2 Fig. 2 illustrates an initially mis-assembled BAC clone from chromosome Y that could be assembled correctly with careful editing. The second step was gap closure. Because gaps tended to be enriched for problematic sequences, gap closure was challenging; it often required multiple attempts using a variety of alternative methods. Gaps were classified into two types: ‘spanned’ and ‘unspanned’. Spanned gaps were those for which the two flanking contig ends were linked by an end-sequenced plasmid. Most such gaps could be closed by primer-directed sequencing of the plasmid, serially extending the |