1. PSEUDOPOTENTIALS
Reference (a bit old but explains how they are generated:
Bachelet, Hamann, Schluter, Phys Rev B 26, 4199 (1982)
What is the difference between the "old" nonlocal pseudopotentials and
the "new" ultrasoft pseudopotentials? What are the energy cutoffs
associated with each?
COARSE MEDIUM FINE
H_00.recpot 400 600 800
H_00.usp 200 270 340
Si_00.recpot 125 200 300
Si_00.usp 80 120 160
O_00.recpot 330 450 500
O_01.recpot 600 750 900
O_02.recpot 620 780 900
O_03.recpot 620 780 900
O_00.usp 260 300 340
COMMENTS IN THE H_00.recpot FILE:
h2001 - very old..
This is a pure (1/r) potential of Hydrogen. It comes from many people's
experience that using an pseudopotential for H do not necessarily
have real advantage.
Convergence testing (5x5x5 A box, LDA).
Eunrel refers to 0.765 bond length (ADF result).
===================================
Ecut E unrel Erel Bond
200 -29.67023 -29.70076 0.813
400 -30.45969 -30.48472 0.804
600 -30.71480 -30.72192 0.787
800 -30.82470 -30.82791 0.780
1000 -30.88436 -30.88717 0.779
1200 -30.92240 -30.92518 0.779
COMMENTS IN THE H_00.usp FILE:
Ultrasoft potential generated using the setting
suggested by Prof. Lee group (H_mhl_01).
H2 dimer, orthorombic cell, a=6.05, b=5.95, c=6.00 Angstrom
Fractional coordinates:
(0.612294 0.622585 0.617397) and (0.544730 0.553885 0.549270)
=============================================================
Ecut Etot dE Force on atom 1
(eV) (eV) (eV/atom) (eV/A)
-------------------------------------------------------------
200 (COARSE) -30.540 0.324 1.18260 1.12637 1.08127
220 -30.652 0.268 1.19001 1.15062 1.19479
270 (MEDIUM) -30.805 0.191 1.09293 1.08480 1.09594
280 -30.846 0.171 1.09470 1.09690 1.09281
320 -30.932 0.128 1.11031 1.11356 1.11282
340 (FINE) -30.965 0.111 1.11338 1.11467 1.10942
380 (PRECISE) -31.032 0.078 1.13022 1.13198 1.12783
400 -31.048 0.070 1.13129 1.13361 1.13356
450 -31.097 0.045 1.13282 1.13608 1.13237
800 -31.187 1.06065 1.06116 1.06096
=============================================================
Validation test
---------------
#1 H2 dimer, exp. bond length 0.7414, CASTEP (GGA, PRECISE)
gives 0.7422 (+0.1%)
2. Choice of Supercell
Build a model for the Si(100) surface.
First, load the bulk Si model:
load model
Cerius2-Models
semiconductors
Si.msi
For an accurate structure, you would then do a geometry
optimization on the bulk, allowing the cell parameters to
vary so as to optimize the Si-Si bond length.
The cell parameter that results from a geometry optimization
of bulk Si using GGA and a 500 eV cutoff is 5.395693 Angstroms.
Next, build a slab from the bulk:
click on surface builder
specify 100
click on the plus sign to get a new model window
now when you click "cleave" you will see the result
you can click "cleave" over and over until you like what you get
How many layers to you want to have in the slab?
Are you going to have to use hydrogens to terminate dangling bonds?
(How many layers are you going to keep "frozen" in the bulk positions?
How many Si atoms do you want in each layer?
How much vaccuum will you need between slabs?
By default, the supercell here has only 1 Si atom per layer
If you are going to want to build the reconstructed Si(100)-(2x1)
surface (or other periodicities) you will probably want more Si
atoms per layer. To build a supercell that will contain one Si
dimer, try the following:
specify 6 A depth (gives 5 Si layers)
specify (1,2) for the surface cell display range
click non-periodic superstructure
switch to crystal builder
click build crystal
alter the cell parameters so that bond
lengths between Si atoms in neighboring
cells are identical to bond lengths between
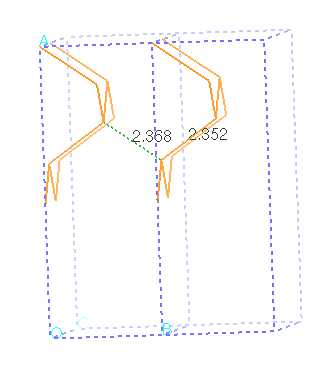
Si atoms in the same supercell (use visualization)
Use the measuring tool to figure out what cell parameter
to specify for "a" in order to get the amount of vaccuum
you want - generally 6-9 Angstroms is enough.
|
Dept. of Chemistry, University of Pittsburgh,
219 Parkman Avenue, Pittsburgh, PA 15260
Phone: (412) 624-8690 FAX: (412) 624-8611 email: jordan at pitt.edu
This page last updated: